|
||||||||||||
 |
 |
 |
 |
 |
 |
 |
 |
| 购买进口仪器、试剂和耗材——就在始于2001年的毕特博生物 www.bitebo.com |
lonza 特殊 无血清 培养基http://bitebo.com/a/gb2312/gongsixinxi/shichanghuodong/2013/0331/5024.html 品牌现货促销 http://www.bitebo.com/plus/list.php?tid=79
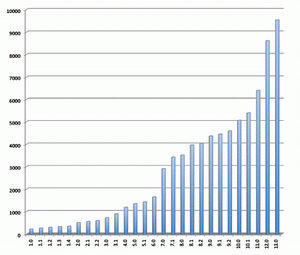
2009年9月15日,Sanger microRNA序列数据库(miRBase)(http://www.mirbase.org/)升级至14.0版。新增1580条miR序列。至此,miRBase中的microRNA序列信息已超过10,000条。14.0版本共收录10,581 条成熟miRNA序列信息,通过验证的microRNA发夹前体从9,539条增至10,883条,共涵盖115个物种。收录于miRNA序列数据库的 miRNA序列数量越来越多(见图),有关miRNA研究的文章和出版物也层出不穷,由此表明,miRNA的研究已经成为目前生命科学领域最重要的研究课题之一。
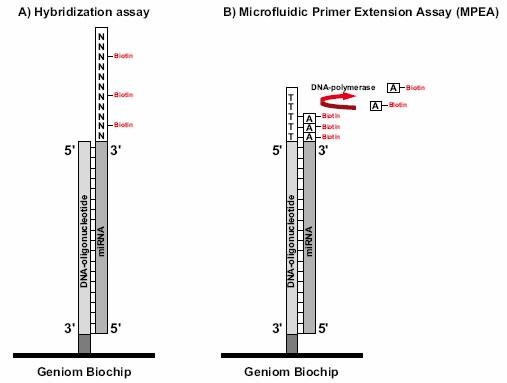
发行于不同版本miRNA数据库的miRNA的数量变化示意图 更为重要的是科研工作者的研究必须与迅速增长的microRNA数据库保持一致,这是确保研究者不错失相关信息的关键。获得miRBase最新数据更新是确保完成miRNA表达谱系分析的唯一途径。对生物标记物研究来讲,评估单个miRNA起着重要的作用。在疾病诊断方面,进行miRNA独特生物标记的多重分析,对于疾病(例如癌症)的分类或预后有重要意义。 然而,一个单独的生物标记物往往在特异性及灵敏度方面受限,而miRNA表达谱具有高特异性,因此它可以反映肿瘤的进化谱系和分化。这样,为了将 miRNA作为诊断标记,有必要对其进行多样性分析。由于高通量技术的使用例如微点阵分析,能够区分单个miRNA,并可以高度精确性地完成miRNA标签信息的收集。 最近,与德国癌症研究中心(DKFZ)合作的,德国Febit公司通过应用Geniom生物芯片系统,发现在胰腺癌病例存在有miRNA生物标记物(和文献报道一致)。应用微点阵观察结果显示,与从较老版本的miRNA序列数据库获取资料相比,使用升级版本数据库(12.0版本代替11.0版本)能够多获得25%的信息。在借助生物标记物来评估胰腺癌的过程中,使用了新的miRNA,而这种miRNA只能从12.0版本的miRNA序列数据库获得。这也说明生物医药应用与不断增长的数据资料保持一致的重要性。 Geniom生物芯片提供研究者最新的miRNA生物芯片。微流体微点阵系统,具备灵活性以迅速地适应新的序列信息。基于最新数据资料,最佳化的捕获探针的合成,是直接在生物芯片的微通道内完成的,从而保证了芯片的生产和最新microRNA同步的独一无二优势。吉奥生物和德国Febit联合推出了和Sanger miRBase 14.0同步的miRNA表达谱芯片。 如果需要,Geniom生物芯片探针能够很容易地根据研究人员的需要而定制。样本检测所需时间短,能够迅速提供可以直接用于发表的数据,因此在竞争激烈的的生物医药研究领域,具有不可替代的地位。 什么是miRNA(microRNA)? miRNAs是一类内源性、约22个核苷酸长度的非编码RNA,具有稳定的特异性序列以调节基因表达。2001年,miRNAs作为线虫生长发育过程中的调节器而被发现,在病毒、植物和动物体中,miRNAs已被公认为主要的调控基因家族之一,通过mRNA靶向作用降解或抑制其翻译。 与蛋白编码基因谱相比,用miRNA表达模式来划分癌症类型的可靠性出乎意料(Lu et al.,2005;Rosenfeld et al.,2008)。此外,在常规收集的、福尔马林固定石蜡包埋(FFPE)的临床组织标本中,miRNAs的表达具有特异性和稳定性,更进一步证明其作为诊断性生物标记的巨大潜能(Li et al., 2007)。 在哺乳动物基因组中,约有70%的miRNA基因定位于特定转录单位。他们经常存在于蛋白编码基因的内含子处,并且优先排列在与其具有相同靶 mRNA的结构处(Kim and Nam,2006)。另一种情况,miRNA基因可能定位于非编码转录单位的内含子或外显子处。组织特异的miRNA表达的调控至今没有完全阐明。 然而,目前的研究清楚地显示miRNAs表达与多种人类恶性肿瘤相关,提示它们代表着一种新型癌症生物标记信号。在特定癌症中找出相关的miRNA,能够提供给我们有用的诊断信息,从而为疾病分类,以及制定出可行的治疗方案打下基础。 产生机制及分子学功能 miRNA下调基因表达发生在转录后水平(Liu et al., 2008)。miRNP复合物与特异的mRNA通过碱基配对相互作用,其二者的结合位点在mRNA的3’端非翻译区,他们的Ago结合蛋白就沉积在 mRNA靶点处。miRNA通过去稳定作用或者核内裂解mRNA介导翻译阻抑,这种阻抑作用依赖于miRNA与靶mRNA之间的互补性。miRNA确切的分子功能也依赖于Ago蛋白动员。 如果与Ago2蛋白结合的成熟miRNA,与靶mRNA完全互补结合,则引起靶mRNA的降解(在植物中比较常见)。mRNA裂解片段通过细胞旁路被清除,而剩下完整的miRNA。靶mRNA的分裂是miRNA调节的优化机制(Jones-Rhoades et al., 2006),动物miRNAs有减退靶蛋白水平的趋势而无核内裂解mRNA(Filipowicz et al., 2005)。另外,成熟的miRNA与只有部分互补序列的靶mRNA结合,随后的翻译抑制可能发生在起始、延长或终止阶段,但是详细机制仍待阐明(Wu and Belasco, 2008)。由于完全互补的靶结合能力不同,每个miRNA可以结合于多种不同的靶基因发挥不同的功能,从而调节多种编码基因。详细的miRNA靶点预测仍然是一个挑战,但是已有许多生物信息手段应用于成熟miRNA序列的前2-8个核苷酸即所谓的“miRNA种子”来进行预测。目前,Vasudevan 等人指出,多种miRNAs不仅能抑制翻译,而且也能增强翻译,是抑制还是增强翻译依赖于细胞周期所处状态。 miRNAs在癌症中起重要作用 miRNAs充当基因调节器作用,能够调控细胞生长、分化和凋亡。在许多人类肿瘤病例中,都发现miRNA表达异常,要么是在成熟的 miRNAs,要么是在前体miRNAs,或者二者皆有。因此,miRNAs表达水平的改变在肿瘤形成中也起重要作用。与正常组织相比,虽然在肿瘤组织中大多数miRNAs表达都下降,但是也有高表达的miRNAs被发现。鉴于此,miRNA基因被认为既充当肿瘤抑制基因也充当癌基因角色(Zhang et al., 2007)。 例如,研究发现肺癌病人的miRNAs let-7表达水平降低(Takamizawa et al., 2004)。在动物体内,let-7的表达依赖于动物的生长发育周期,在发育早期let-7低表达,而分化成熟的组织let-7高表达。下调let-7的表达水平致使分化失败,从而导致了癌症的发生。另外,实验表明,RAS癌基因是miRNA let-7的直接靶点,说明在肺癌发生过程中,let-7通过调节RAS的表达发挥抑癌基因作用。第二种情况,通过作用于抗凋亡基因BCL2靶点,下调 miR-15和miR-16在慢性淋巴瘤的表达。另一方面,致癌的miRNAs通过负调控抑制抑癌基因或调节细胞分化或细胞凋亡,从而促进肿瘤生长。例如,miR-17-92在许多癌症如肺癌或淋巴瘤的表达明显增加,它的形成可以增加肺癌细胞的生长。进一步研究表明,到目前为止,参与肿瘤形成的 miRNAs中,至少50%的miRNAs基因定位于癌症相关基因组区域(Calin et al., 2004)。 通过研究miRNAs表达谱来进行癌症分类 基于miRNAs表达谱研究,我们可以就肿瘤发育形成和分化程度对其进行分类。较传统的基于mRNA表达研究的分类方法,具有更高的精确度(Lu et al., 2005; Rosenfeld et al., 2008)。在包含多种人类癌症的系统性研究中,几乎所有的miRNAs都显示出不同的表达,这使得将其用以鉴定不同发育起源的肿瘤成为可能。这种人类癌症的分类方法,通过测定相关的大约200条小数量miRNAs的表达来完成,而如果通过mRNA基因表达方法的话,即使使用几千个mRNA也不能得到同样结果。另外研究显示,miRNAs的表达与特异的病理学特点如肿瘤期或增殖指数具有强相关性。相比之下,由于mRNA转录后修饰发生在从DNA转变成蛋白质的过程中,因此,其表达谱常常不能直接得出生物学或者具有临床意义的结果,而miRNAs能较为直接的反映基因功能水平。 使用miRNAs为肿瘤分类的另外一个巨大益处:将大大改善临床研究中常规收集保存的,通过福尔马林固定的石蜡包埋(FFPE)组织的稳定性。较长的mRNAs在甲醛固定和加工处理期间最可能被降解和化学修饰。而miRNAs很小,蛋白质又受RISC络合物的保护,故其固定方法较持久,能够长期保持。通过广泛的Snap Frozen Cells研究发现,使用FFPE样本做miRNAs表达谱具有很高的可靠性(Li et al., 2007)。 miRNAs是诊断疾病和判断预后的稳定和可靠的生物标记物 miRNAs除了显著的组织特异性外,还具有诊断癌症和判断预后的作用。由于它们的基因调控灵活,将miRNAs用于癌症治疗的潜力显而易见。所谓的抗- miRNAs寡核苷酸类物质(AMOs),与致癌的miRNAs互补,能够特异抑制肿瘤组织中miRNAs的活性。另一方面,起肿瘤抑制基因作用的 miRNAs的高表达可能对肿瘤的治疗有益。 miRNAs不仅为癌症,也为许多其他疾病如病毒感染性疾病、心血管疾病等提供可行的治疗方案,在这些疾病治疗中也涉及miRNAs的基因调控。 对miRNAs基因调控的研究是诸多研究的焦点,有关miRNAs自身基因表达调控机制的知识仍待进一步扩展。另外,miRNAs被认为受控于渐成机制,而不是仅仅局限于自身组织和肿瘤表达模式。实际上,若干miRNAs被DNA甲基化调控。应用去甲基化处理治疗人类膀胱癌,Saito et al.发现大约5%的人类miRNAs的表达水平提高了至少了3倍。最显著的效应是miR-127,其对应的基因嵌入CpG岛。通过表观遗传学方法复性后,其靶基因之一卟啉-癌基因BCL6的表达是下调的,从而得出miR-127可能发挥肿瘤抑制基因的作用。这样,通过表观遗传学途径来进行抗癌治疗将变得可行。 对miRNAs表达水平高通量分析的最普遍的方法是使用寡核苷酸微阵列(Liu,C.-G. et al., 2004), 在大样本的研究中,使用这种方法能够同时测量成百上千miRNAs的基因表达。另外一种微阵列方法叫作微流引物延伸检测Microfluidic Primer Extension Assay(MPEA),这种检测方法以Febit Geniom®微阵技术的使用为基础,miRNA在高度灵敏的微阵列杂交前,不需要标记。接着,DNA聚合酶I的Klenow片段直接加入微量流动芯片的通道中,相应的miRNA得以特异延伸。此方法将杂交检测的特异性与酶延伸的高辨别能力相结合(Vorwerk et al., 2008)。
MPEA相对于现有的其它微阵列方法,显示出突出优势。由于miRNA不需经过富集、PCR扩增或标记等预处理而直接导入,这样就确保避免了实验误差的产生(见图, A为常规杂交方法,B为微流引物延伸)。传统的杂交检测最适合鉴别杂交靶点中心位点上的错配,相比之下,MPEA提供了更高水平的灵敏度,由于酶催化的延伸仅在3’末端几乎完全配对时才会发生,因此很少会出现交叉杂交信号。与传统的RNA-预处理,Klenow延伸列阵检测(RAKE)相比,MPEA也显示出几点主要优势(Nelson et al., 2004)。RAKE微数列通过与其相应的寡核苷酸的5' 末端结合于表面,而MPEA阵列的寡聚核苷酸捕获探针与其3' 末端相连。MPEA的这种结合方式不仅排除了探针的自身延长,尤其重要的是说明这种延长作用远离微通道管腔表面而没有任何位阻。由于微流体通道的使用,也大大降低了所需RNA样本量。MPEA具有高度的灵敏性,无需扩增,就足以检测出来源于福尔马林固定样本,组织针孔吸取样本或激光捕获显微分离样本的纳克级的总RNA。 |
购买进口仪器、试剂和耗材——就在始于2001年的毕特博生物
www.bitebo.com |
|